
![]()
БРЕМЯ ЗАБОЛЕВАНИЯ
Болезнь Фабри — это прогрессирующее, угрожающее жизни заболевание с полиорганным поражением, которое относится к группе лизосомных болезней накопления и вызывается мутациями гена GLA.
Болезнь Фабри — это редкое заболевание с полиорганным поражением, которое относится к группе лизосомных болезней накопления и вызывается мутациями гена GLA, расположенного на X-хромосоме. Ген GLA кодирует фермент альфагалактозидазу A (А-ГАЛ A)1. Выявлено более 1000 мутаций гена GLA, вызывающих снижение уровня активности фермента А-ГАЛ А (см. «Генетическое наследование болезни Фабри»)2–5. У пациентов с болезнью Фабри наблюдается нарушение активности А-ГАЛ А, которое ведет к накоплению гликосфинголипидов — глоботриаозилцерамида (Gb3) и глоботриаозилсфингозина (лизо-(Gb3)) почти во всех видах клеток и в разных органах (рис. 1)4,6. В результате прогрессирующего накопления гликосфинголипидов в тканях различных органов, развиваются клинические проявления болезни Фабри (рис. 2)4,5. По уровню активности А-ГАЛ А можно спрогнозировать фенотип заболевания, а именно классическую форму или форму с поздним началом4. Мутации гена GLA, которые связаны с практически полным отсутствием активности указанного фермента (<1%), наблюдаются при классической форме болезни Фабри, тогда как сохранение остаточной активности А-ГАЛ А (≥1‒30%) обычно сопровождается развитием формы с поздним началом1,7–9. Классический фенотип болезни Фабри имеет, как правило, характерную клиническую картину, а при фенотипе с поздним началом течение заболевания может быть разным, в некоторых случаях даже менее тяжелым, и клинические проявления могут ограничиваться только одним органом10,11.
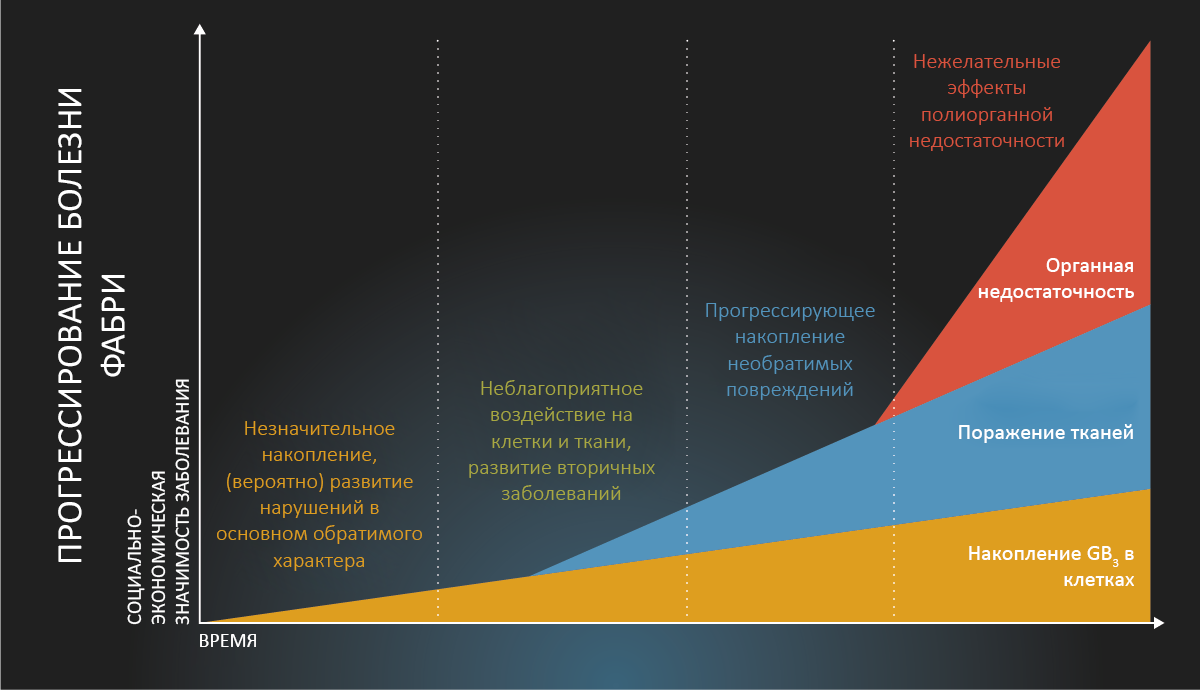
Рис. 1.
Прогрессирование болезни Фабри. Воспроизведено с разрешения Eng CM et al. J Inherit Metab Dis 2007; 30: 184-19212.
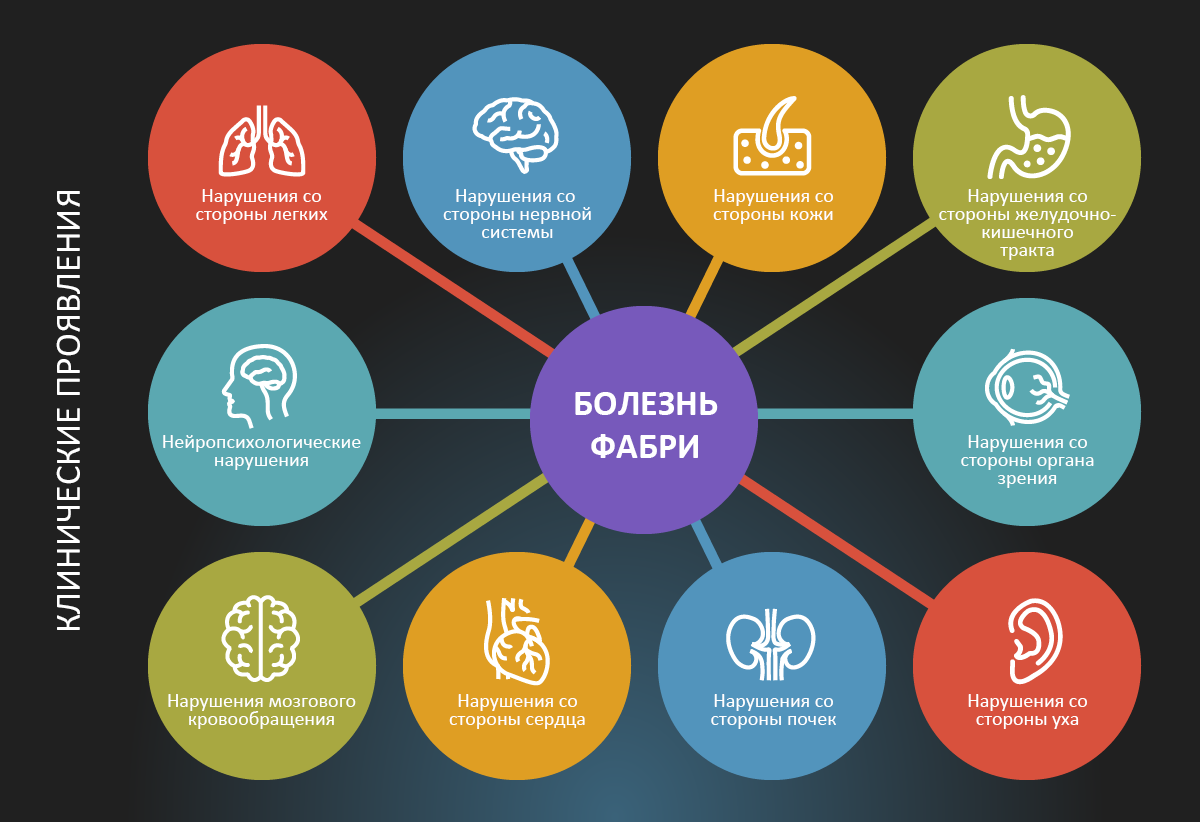
Рис. 2.
Клинические проявления классической формы болезни Фабри разнообразны7.
По данным литературы, болезнь Фабри встречается с частотой приблизительно 1 случай на 40 000 мужского населения и приблизительно 1 случай на 20 000 женского населения11,13. Однако данные программ скрининга новорожденных свидетельствуют о том, что распространенность болезни Фабри может быть существенно выше, чем предполагалось ранее11,13–19. Болезнь Фабри может встречаться у представителей всех этнических, расовых и демографических групп20.
Поскольку болезнь Фабри характеризуется тяжелым полиорганным поражением и широким спектром признаков и симптомов, по мере прогрессирования она может привести к развитию недостаточности жизненно важных органов и преждевременной смерти пациента11,21. Кроме того, возраст возникновения первых проявлений болезни Фабри у мужчин и женщин может различаться21. Таким образом, болезнь Фабри может оказывать существенное влияние на состояние пациентов, во многих случаях снижая качество их жизни22.
- Vardarli I, Rischpler C, Herrmann K, et al. Diagnosis and screening of patients with Fabry disease. Ther Clin Risk Manag 2020; 16: 551-558.
- McCafferty EH, Scott LJ. Migalastat: a review in Fabry disease. Drugs 2019; 79: 543-554.
- Tuttolomondo A, Simonetta I, Duro G, et al. Inter-familial and intra-familial phenotypic variability in three Sicilian families with Anderson-Fabry disease. Oncotarget 2017; 8: 61415-61424.
- Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2017; 91: 284-293.
- Felis A, Whitlow M, Kraus A, et al. Current and investigational therapeutics for Fabry disease. Kidney Int Rep 2019; 5: 407-413.
- Brady RO, Gal AE, Bradley RM, et al. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med 1967; 276: 1163-1167.
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 2018; 123: 416-427.
- Clarke JT. Narrative review: Fabry disease. Ann Intern Med 2007; 146: 425-433.
- Michaud M, Mauhin W, Belmatoug N, et al. When and how to diagnose Fabry disease in clinical pratice. Am J Med Sci 2020; 360: 641-649.
- Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol 2017; 28: 1631-1641.
- Desnick RJ, Ioannou YA, Eng CM. α-galactosidase A deficiency: Fabry disease. In: Scriver C, Beaudet A, Sly W, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. 8th Edition. New York, NY: McGraw-Hill, 2001.
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis 2007; 30: 184-192.
- Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns 2008; 17: 79-83.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet 2006; 79: 31-40.
- Colon C, Ortolano S, Melcon-Crespo C, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr 2017; 176: 1075-1081.
- Inoue T, Hattori K, Ihara K, et al. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet 2013; 58: 548-552.
- van der Tol L, Smid BE, Poorthuis BJHM, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet 2014; 51: 1-9.
- Scott CR, Elliott S, Buroker N, et al. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J Pediatr 2013; 163: 498-503.
- Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 2012; 379: 335-341.
- Mehta A, Hughes DA. Fabry disease. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle, 2002.
- Mehta A, Clarke JTR, Giugliani R, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet 2009; 46: 548-552.
- Arends M, Hollak CEM, Biegstraaten M. Quality of life in patients with Fabry disease: a systematic review of the literature. Orphanet J Rare Dis 2015; 10: 77.
Дифференциальная диагностика
Генетический анализ и анализ родословной с диагностической целью
После установления диагноза болезни Фабри в каждом случае рекомендуется провести тщательный анализ родословной пациента, чтобы выявить других членов семьи с риском заболевания.
Дифференциальная диагностика
Нефрологические аспекты заболевания
Почечные проявления могут возникать уже на ранних стадиях заболевания, поэтому нефрологам следует знать о возможности наличия у пациента болезни Фабри.
Дифференциальная диагностика
Кардиологические аспекты заболевания
В связи с высокой распространенностью поражения сердца у пациентов с болезнью Фабри кардиологи играют важную роль в скрининге и диагностике заболевания. Кардиологам рекомендуется сохранять настороженность в отношении болезни Фабри.
Дифференциальная диагностика
Неврологические аспекты заболевания
Неврологи могут помочь в ранней диагностике болезни Фабри и выявить поражения нервной системы, связанные с заболеванием.
БРЕМЯ ЗАБОЛЕВАНИЯ
Болезнь Фабри
Что такое болезнь Фабри?
Болезнь Фабри — это X-сцепленное генетическое заболевание, вызываемое мутациями в гене GLA, который кодирует лизосомный фермент А-ГАЛ А.